Chapitre 1 Métagénomique
Quel est le point commun entre une poignée de terre, du camembert et votre intestin ? Tous hébergent une communauté de micro-organismes –bactéries, virus, champignons…– collectivement appelés le microbiote. Aussi extrêmes soient-ils, tous les environnements hébergent des communautés microbiennes. Comme l’on pourrait s’y attendre, les bactéries présentes dans le tube digestif d’escargots sous-marins (Aronson, Zellmer, & Goffredi, 2017) ne sont pas les mêmes que celles présentes dans le désert d’Atacama (Araya, González, Cardinale, Schnell, & Stoll, 2020), mais même au sein d’environnements comparables, il existe une grande variabilité tant au niveau des espèces présentes que de leurs abondances.
Depuis la fin des années 2000, le développement des techniques de séquençage haut-débit et la baisse de leurs coûts (voir figure 1.1) ont rendu accessible l’ensemble des génomes du microbiote, aussi appelé microbiome ou métagénome.
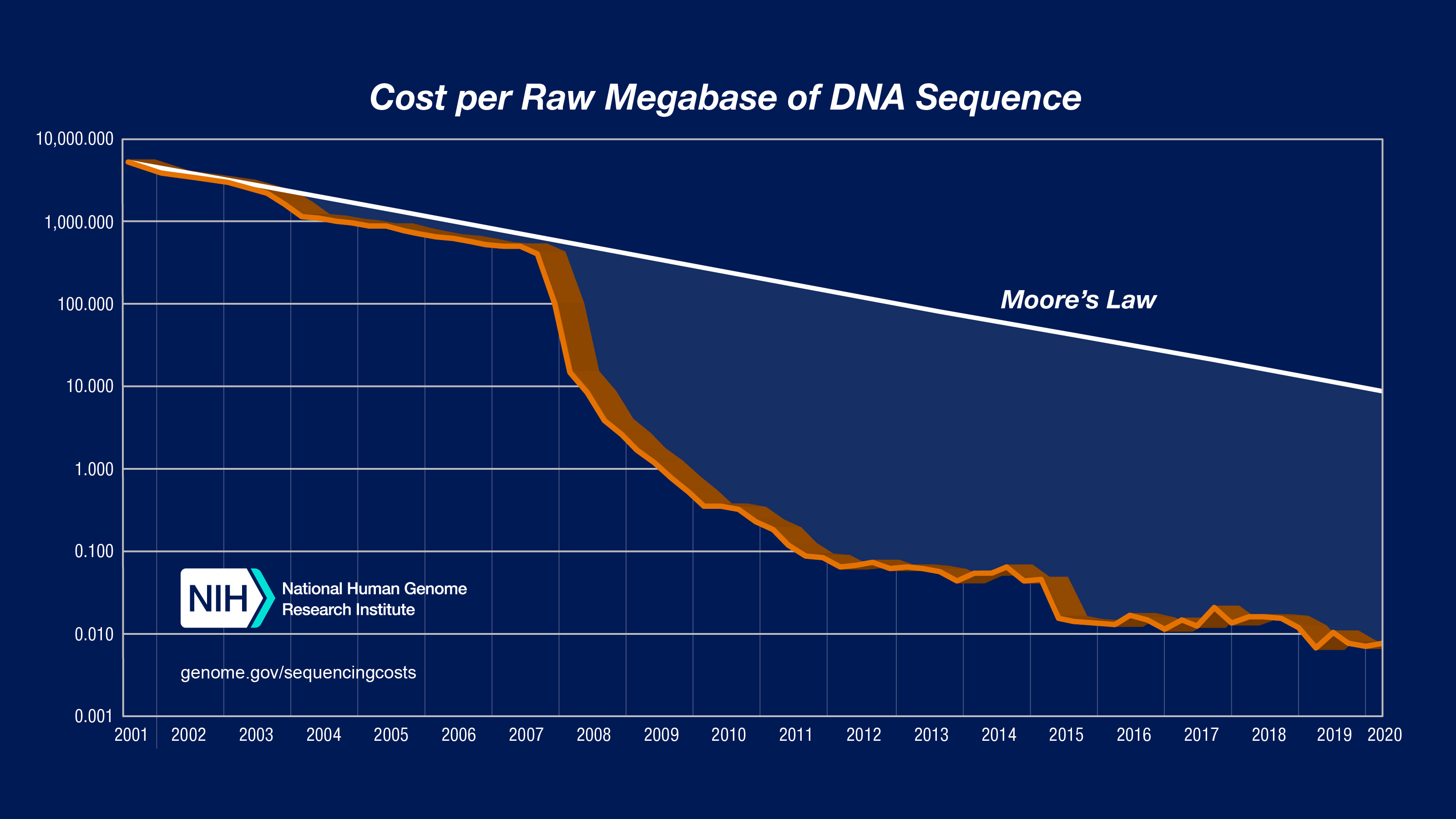
Figure 1.1: Évolution du coût de séquençage (en dollar) d’une mégabase d’ADN, en échelle logarithmique, tiré de www.genome.gov.
1.1 Le microbiote intestinal humain
Même si les développements méthodologiques présentés dans cette thèse ont une portée générale et ne sont pas limités à un type de microbiote en particulier, ils sont effectués avec le microbiote intestinal humain en ligne de mire. Nous présentons ici quelques propriétés de ce microbiote afin que le lecteur prenne conscience de son importance et ait un exemple concret auquel se raccrocher par la suite.
1.1.1 Description
Le tractus gastro-intestinal, ou tube digestif, abrite une communauté microbienne composée d’environ cent mille milliards de micro-organismes pour un poids d’environ \(2~\text{kg}\) (Ley, Peterson, & Gordon, 2006). Par abus de langage, on l’appelle le microbiote intestinal. Chez les adultes au mode de vie occidental et en bonne santé, les bactéries appartenant à l’embranchement (phylum) des Firmicutes représentent plus de \(60~\%\) de la composante bactérienne du microbiote. Si l’on y ajoute celles appartenant aux embranchements des Actinobacteria et des Bacteroidetes, cette fraction monte à \(90~\%\) (Zhernakova et al., 2016).
La variabilité inter-individus reflète principalement des facteurs environnementaux ou comportementaux, qui structurent fortement les flores microbiennes, notamment: le régime alimentaire (David et al., 2014), l’âge (O’Toole & Claesson, 2010; Yatsunenko et al., 2012) mais aussi la prise d’antibiotiques (Bokulich et al., 2016; Palleja et al., 2018), la présence d’un animal de compagnie (Kates et al., 2020), etc.
1.1.2 Rôle
Les micro-organismes qui colonisent le tractus gastro-intestinal dégradent les glucides qui n’ont pas été préalablement absorbés par l’hôte (Flint, Scott, Duncan, Louis, & Forano, 2012; Rowland et al., 2018). Ces réactions produisent des acides gras à chaîne courte (AGCC) qui sont des sources d’énergies importantes pour l’humain. Plusieurs voies métaboliques, présentes chez différentes bactéries, permettent de produire des AGCC et il n’existe donc pas un profil unique pour tous les humains. Des études à grande échelles (Arumugam et al., 2011) ont cependant permis de dégager des « profils types », appelés entérotypes, caractérisés entre autres par l’alimentation.
Certaines dégradations de sucres effectuées par le microbiote sont inaccessibles aux seules voies métaboliques humaines : le microbiote constitue donc un compagnon indispensable pour assimiler pleinement les nutriments que nous consommons. Citons l’exemple de la population japonaise, qui consomme en moyenne \(14.2~\text{g}\) de nori (un type d’algue) par jour et dans laquelle les enzymes porphyranases et agarases responsables de la dégradation des algues sont produites par la bactérie Bacteroides plebeius. Cette capacité a été obtenue à la suite d’un transfert horizontal de gène de la part Zobellia galactanivorans, une bactérie marine (Hehemann et al., 2010).
Le système immunitaire doit également beaucoup au microbiote intestinal (Blander, Longman, Iliev, Sonnenberg, & Artis, 2017). Parmi les AGCC précédemment cités, on retrouve le butyrate, un métabolite ayant des propriétés anti-inflammatoires et favorisant la prolifération cellulaire au sein de la muqueuse intestinale, ce qui participe à la prévention du cancer colorectal (Canani et al., 2011). La composition du microbiote intestinal a également un effet sur l’efficacité des vaccins (Valdez, Brown, & Finlay, 2014). Eloe-Fadrosh et al. (2013) ont mis en évidence que les patients les plus répondeurs pour un vaccin antityphoïdique étaient ceux présentant une proportion plus importante de Clostridiales. Mentionnons enfin dans ce domaine, la bactérie Bacteroides thetaiotamicron qui induit une production du peptide antimicrobien \(\text{LL-37}\), lequel protège à son tour son hôte contre les infections par Candida albicans (Fan et al., 2015).
La signalisation biochimique bidirectionnelle entre le tractus gastro-intestinal et le système nerveux central, communément appelée axe intestin-cerveau, est grandement affectée par le microbiote intestinal. Les résultats les plus spectaculaires sont obtenues chez la drosophile, dans laquelle le microbiote intestinal participe à la modulation des comportements locomoteur (Schretter et al., 2018) et sexuel (Sharon et al., 2010), et chez le rat, dans lequel la production d’un métabolite (l’indole) par certaines bactéries du microbiote induit des troubles du comportement et des troubles de l’anxiété (Jaglin et al., 2018).
1.1.3 Dysbioses
Le terme dysbiose désigne un déséquilibre du microbiote, qui se traduit généralement par une perte de diversité ou par la surreprésentation d’une espèce. La table 1.1 présente un ensemble de maladies qui seraient causées par ou associées à une dysbiose.
| Type de maladie | Maladie |
|---|---|
| Maladies métaboliques | Obésité (Turnbaugh et al., 2009)
Diabète de type 2 (Qin et al., 2012) Cirrhose (Qin et al., 2014) |
| Maladies immunitaires | Maladie de Crohn (Morgan et al., 2012)
Syndrome de l’intestin irritable (Chong et al., 2019) Sclérose en plaques (Cekanaviciute et al., 2017) Asthme (Stokholm et al., 2018) |
| Maladies psychiatrique | Dépression (Foster & Neufeld, 2013)
Schizophrénie (Zheng et al., 2019) |
| Maladies neurologiques | Maladie d’Alzheimer (Pistollato et al., 2016)
Maladie de Parkinson (Bedarf et al., 2017) Syndrome de Gilles de La Tourette (Ding et al., 2019) |
| Autres maladies | Eczema (Abrahamsson et al., 2012)
Maladies cardiovasculaires (Kelly et al., 2016) Cancer colorectal (Zeller et al., 2014) Entérocolite nécrosante (Mai et al., 2011) |
1.1.4 Utilisations
Les liens entre microbiote et santé étant très nombreux, la recherche académique ainsi que les départements de recherche et innovation des industries pharmaceutiques et agroalimentaires n’ont pas attendu pour se lancer dans la recherche d’applications et de traitements tirant parti de ces micro-organismes.
Si certaines bactéries sont bénéfiques pour l’organisme, pourquoi ne pas augmenter volontairement leur quantité dans le microbiote ? C’est ce que proposent les approches probiotiques –aussi appelés bioaugmentation– qui consistent à administrer (le plus souvent par voie orale) des bactéries vivantes et non-pathogènes (Gibson et al., 2017). Les probiotiques sont majoritairement considérés comme des compléments alimentaires, qui n’ont pas besoin de montrer leur efficacité pour être commercialisés. Des études cliniques prouvent cependant leur efficacité dans certains cas, comme l’utilisation d’un probiotique à base d’une souche de Bifidobacterium longum pour diminuer la dépression chez les patients souffrant du syndrome de l’intestin irritable (Pinto-Sanchez et al., 2017).
Plutôt que de fournir directement des souches vivantes, l’approche par prébiotiques fournit des nutriments non digestibles par l’hôte mais stimulant la croissance de certaines bactéries (Gibson et al., 2017). Si les aliments « riches en fibres » peuvent être considérés comme des prébiotiques, des prébiotiques de synthèse font leur apparition sur le marché. Nestlé a par exemple déposé un brevet sur des prébiotiques à base d’oligosaccharides qui réduisent la présence de Streptococcus chez l’enfant dans le but de diminuer le risque d’obésité une fois adulte (Sakwinska, Berger, Zolezzi, & Holbrook, 2017). Dans le même registre, la baguette « Amibiote » (contraction entre ami et microbiote), issue d’un partenariat entre INRAE et Bridor, contient \(11~\text{g}\) de fibres pour \(100~\text{g}\) de pain (contre \(2.9~\text{g}\) pour une baguette normale) et favorise la croissance de trois bactéries probiotiques.
Pour modifier la composition du microbiote intestinal, la méthode la plus efficace reste la transplantation fécale. Déjà pratiquée en Chine au \(\text{IV}^{\text{e}}\) siècle (Zhang, Luo, Shi, Fan, & Ji, 2012), la repopulation de l’intestin d’un sujet malade avec le microbiote d’un sujet sain a fait un retour en force en montrant des résultats spectaculaire pour le traitement des infections à Clostridium difficile comparé aux thérapies habituelles (prise d’antibiotiques avec ou sans lavement) (Van Nood et al., 2013). L’infection à Clostridium difficile provoque des diarrhées potentiellement mortelles et se produit chez des patients dont le microbiote intestinal a déjà subi une perte de diversité, laissant la place au pathogène pour se développer.
Si la transplantation fécale pourrait servir de traitement à d’autres maladies, comme le syndrome de Gilles de La Tourette (Ding et al., 2019), ce n’est pas le seul dessein dans lequel l’utilisation de cette technique est possible. Des transplantations fécales autologues peuvent être envisagées dans le cas où une altération du microbiote serait à prévoir. Cette technique a été testée avec succès par la société française MaaT Pharma pour des patients souffrant de leucémie aiguë myéloïde. Leurs selles sont collectées avant la chimiothérapie et une transplantation fécale autologue permet de restaurer un microbiote diversifié comparable à celui présent dans le tractus avant le traitement (Mohty et al., 2018). Une utilisation plus originale en est faite chez les koalas : la transplantation fécale leur permet de diversifier les espèces d’eucalyptus qu’ils sont capables de digérer, ce qui augmente leurs chances de survie alors que leur environnement est menacé (Reardon, 2018).
La médiatisation de ces bons résultats a conduit à l’apparition sur les réseaux sociaux de protocoles pour des transplantations fécales « à faire soi-même » (Ekekezie et al., 2020), y compris pour des indications pour lesquelles elles ne sont (pour l’instant) pas recommandées comme le syndrome de l’intestin irritable ou les troubles de l’autisme. En fonction des pays, la thérapie fécale est considérée comme un médicament ou une transplantation au même titre que les organes.
Plutôt que de modifier le microbiote et d’espérer que la nouvelle composition sera favorable, il est possible d’administrer directement des composés issus des bactéries désirées. C’est sur ce terrain que se place la biotech Enterome, qui a montré que l’administration de peptides issus du microbiote déclenchait une réponse immunitaire capable de s’attaquer à des tumeurs spécifiques (Chene et al., 2019).
Une autre façon de tirer parti du microbiome est de s’en servir comme d’un biomarqueur : regarder sa composition peut être un moyen de déceler une maladie sans avoir recours à des tests invasifs ou plus coûteux. Zeller et al. (2014) ont par exemple proposé un modèle prédictif basé principalement sur les abondances de souches de Fusobacterium nucleatum, Porphyromonas asaccharolytica et Peptostreptococcus stomatis pour détecter de façon précoce un cancer colorectal. Chez les nouveaux-nés, l’entérocolite nécrosante est précédée d’une dysbiose caractéristique (Mai et al., 2011) permettant de l’identifier et de proposer un traitement adapté avant que la maladie ne leur soit fatale.
Toutes ces opportunités ont incité de nombreuses sociétés à se lancer sur le marché du microbiote dans l’espoir de croissances rapides. Mais le réveil peut être douloureux lorsque les espoirs se fracassent sur le mur de la réalité, comme Seres Therapeutics en a fait les frais. Le 29 juillet 2016, l’annonce de l’échec de la phase II de son probiotique SER-109 contre les infections à Clostridium difficile a entraîné une chute du cours de son action près de \(70~\%\) (figure 1.2). L’entreprise a publié un communiqué en janvier 2017 indiquant que cet échec était dû à une erreur de protocole et qu’elle allait par conséquent entamer un essai de phase III, mais cela n’a pas suffi à rassurer les investisseurs.
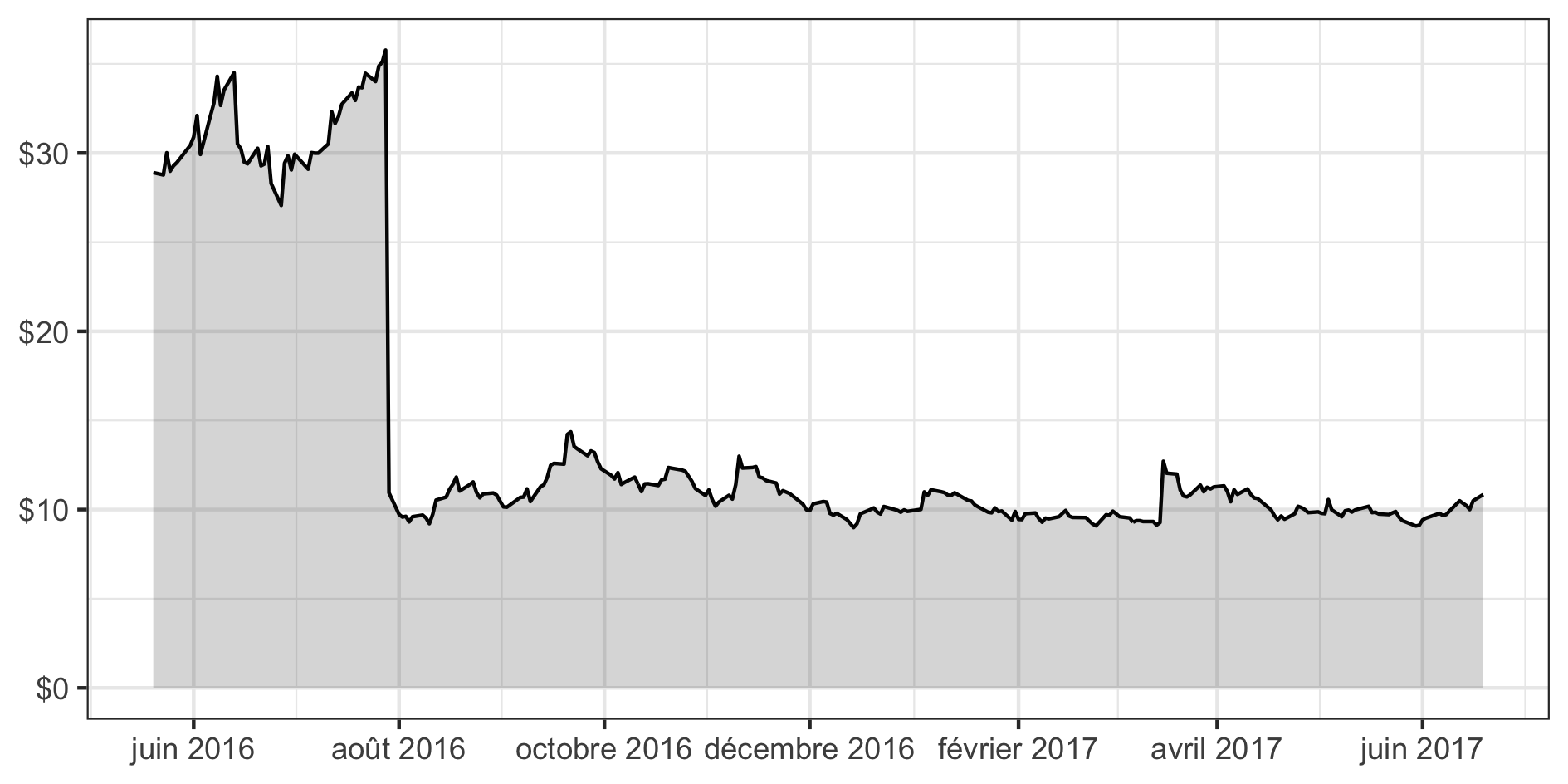
Figure 1.2: Cotation de Seres Therapeutics à partir de l’été 2016.
1.2 Caractérisation du microbiote
Nous décrivons ici brièvement les différentes méthodes et techniques utilisées pour caractériser le microbiote.
1.2.1 Collecte des échantillons
La collecte des échantillons et le mode de conservation des échantillons sont des étapes cruciales pour la reproductibilité des analyses et la fiabilité des résultats. En effet, le temps de stockage avant une congélation à \(-80~^\circ\text{C}\) (Cuthbertson et al., 2014) ou le nombre de dégels successifs (Sergeant, Constantinidou, Cogan, Penn, & Pallen, 2012) peuvent avoir un impact important sur la composition microbienne des échantillons, en affectant certains genres plutôt que d’autres.
Il est donc souhaitable d’avoir les conditions les plus homogènes possibles. Cependant, il peut être relativement compliqué d’obtenir des conditions homogènes de collecte et de conservation, notamment quand il s’agit d’études longitudinales où la récolte d’échantillons s’étale sur plusieurs pays ou plusieurs années, avec des équipes différentes, ou bien quand celle-ci est effectuée au domicile du donneur dans le cadre d’études participatives, telles que l’American Gut Project (McDonald et al., 2018).
1.2.2 Séquençage par gène marqueur
Le séquençage par gène marqueur, en général la sous-unité 16S de l’ARN ribosomique, permet de faire un inventaire taxonomique des espèces présentes dans le microbiote et de répondre à la question « qui est là ? ». Fait notable, le gène codant pour le 16S est présent chez toutes les espèces de bactéries et archées (Kembel, Wu, Eisen, & Green, 2012). Sa séquence présente une succession de régions conservées, idéales pour l’extraire et l’amplifier par PCR, et des régions hypervariables, qui permettent de déterminer à quel genre (et dans une certaine mesure à quelle espèce) il appartient et de reconstruire une phylogénie. Il bénéficie également de bases de données taxonomiques extrêmement riches (par exemple, SILVA (Quast et al., 2012)). Pour toutes ces raisons, le gène 16S est un marqueur idéal pour identifier les différents micro-organismes présents dans un échantillon et quantifier leur abondance (Morgan & Huttenhower, 2012).
Les différentes étapes d’analyse s’enchaînent comme suit. Après extraction de l’ADN, on amplifie le gène 16S par PCR avec des amorces universelles, calibrées à partir des régions conservées, puis on séquence une ou plusieurs régions hypervariables (pour une longueur totale d’environ 550 paires de bases) par séquençage haut débit. Une fois ces portions d’ADN séquencées, on a accès aux séquences brutes des nucléotides qui les composent, appelées lectures ou reads.
Ces lectures doivent ensuite subir un contrôle qualité. En effet, les technologies de séquençage produisent des lectures trop courtes ou de mauvaise qualité (Modolo & Lerat, 2015) qui doivent être éliminées. De plus, lors de l’amplification par PCR, il peut y avoir des évènements d’hybridation entre séquences d’ADN (Meyerhans, Vartanian, & Wain-Hobson, 1990) qui créent des séquences chimériques et augmentent artificiellement la richesse microbienne. Des algorithmes ont été proposés pour identifier et filtrer les séquences chimériques avant la suite des analyses (Edgar, 2016; Wright, Yilmaz, & Noguera, 2012).
Une première approche, dite affiliation first, pour identifier à quelles espèces correspondent les lectures séquencés est de trouver une correspondance entre celles-ci et des séquences de références dans des bases de données de gènes 16S comme RDP (Maidak et al., 2000, 1997), SILVA (Quast et al., 2012) ou Greengenes (DeSantis et al., 2006). Cette méthode est rapide et facilement parallélisable mais rend impossible le regroupement et l’analyse des lectures qui n’ont pas d’homologues dans les bases de référence.
L’approche la plus utilisée, dite clustering first, consiste à rassembler les lectures au sein de groupes appelés OTUs, pour Operational Taxonomic Units. Ce partitionnement se fait en agglomérant toutes les séquences qui ont au moins \(97~\%\) de similarité de séquence. Des outils bioinformatiques comme Mothur (Schloss et al., 2009) ou QIIME (Caporaso et al., 2010) utilisent des algorithmes de classification ascendante hiérarchique pour effectuer ce partitionnement, ce qui est coûteux en calcul et en mémoire. Des algorithmes gloutons (Edgar, 2010) permettent d’accélérer et de réduire l’empreinte mémoire de cette étape de partitionnement. À l’issue de cette étape, un représentant est ensuite choisi pour chaque groupe afin de lui assigner une affiliation taxonomique en comparant ce représentant à des bases de références. Les séquences des représentants permettent également de déterminer un arbre phylogénétique des OTUs. Comparée à l’approche affiliation first, l’identification par OTU a l’avantage de pouvoir gérer des espèces non présentes dans les bases de données de référence en les considérant simplement comme mal affiliées. Le microbiote est finalement résumé par une table de comptage \(X = (x_{ij})\) où \(x_{ij}\) correspond au nombre de lectures de l’OTU \(i\) dans l’échantillon \(j\).
Au delà du partitionnement par similarité de séquences, il existe d’autres méthodes pour partitionner l’ensemble des séquences. Mentionnons par exemple le regroupement aux sein d’ASVs, pour Amplicon Sequence Variants, qui a vocation à reconstruire les séquences exactes des représentants à l’aide d’un modèle probabiliste des erreurs de séquençage (Callahan, McMurdie, & Holmes, 2017; Callahan et al., 2016), ou au sein d’oligotypes, qui se focalisent sur les sites nucléotidiques de grande variabilité (Eren, Borisy, Huse, & Welch, 2014; Eren et al., 2013). Le point de différenciation majeur de ces méthodes par rapport aux OTUs est de ne pas donner le même poids à toutes les positions lors de la construction des groupes de lectures.
La caractérisation par gène marqueur, en particulier le 16S, est bon marché, rapide et s’appuie sur des outils matures. Elle souffre néanmoins de quelques inconvénients :
Lors de l’amplification par PCR, outre la possible création de chimères, les taxons très abondants vont avoir plus de chances de voir leurs deux brins s’apparier entre eux plutôt qu’avec une amorce, ce qui brise la chaîne de réplication (Mathieu-Daudé, Welsh, Vogt, & McClelland, 1996). Les taxons peu présents vont au contraire avoir plus de chances d’aller jusqu’au bout de la chaîne de réplication et leur abondance sera surestimée.
Le nombre de copies du gène 16S varie entre espèces, dans un rapport de \(1\) à \(21\) (Stoddard, Smith, Hein, Roller, & Schmidt, 2015). Utiliser cette information pour corriger la quantification des OTU améliore l’estimation de la composition microbienne, mais le nombre de copies n’est pas toujours disponible (Kembel et al., 2012), en particulier pour les groupes microbiens peu étudiés.
Le 16S est limité à la fraction bactérienne du microbiote. D’autres marqueurs comme l’ITS1 et l’ITS2 doivent être utilisées pour la fraction fongique du microbiote. Ces derniers souffrent également du biais du nombre de copies (mais dans un rapport de \(1\) à \(1~000\)) et de bases de référence nettement moins riches.
Le séquençage par gène marqueur ne permet d’obtenir une résolution taxonomique qu’au niveau du genre, éventuellement de l’espèce dans certains cas favorables. Il ne permet pas non plus de déterminer les fonctions ou voies métaboliques présentes dans le microbiote. Ces dernières peuvent en effet être spécifiques aux souches au sein d’une espèce et nécessitent d’adopter une stratégie non ciblée.
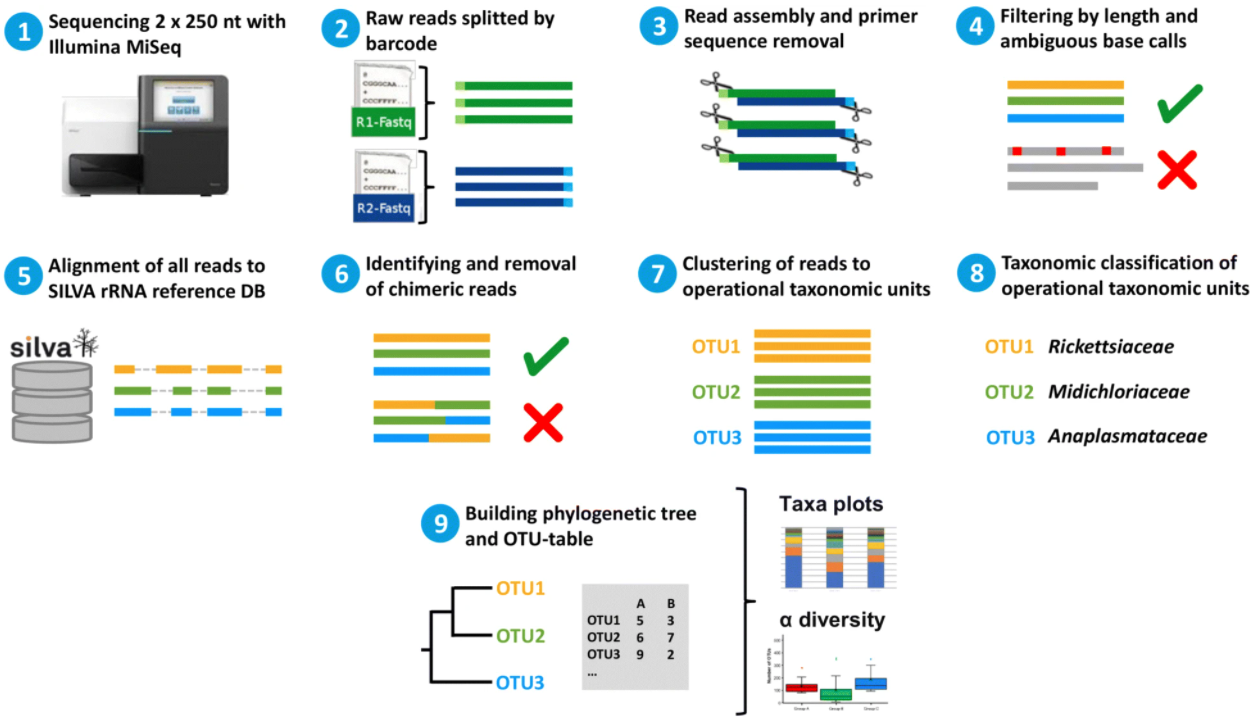
Figure 1.3: Résumé des étapes du séquençage par gène marqueur, tiré de Regier et al. (2019).
1.2.3 Séquençage non ciblé
Comme son nom l’indique, le séquençage non ciblé, ou whole genome shotgun souvent abrégé en shotgun, cible l’ensemble du matériel génétique présent dans les échantillons, et non un unique gène marqueur. Il permet d’étudier le potentiel fonctionnel du microbiote et de répondre à la question « qui peut faire quoi ? ».
On commence par extraire l’ADN contenu dans les cellules puis on le brise par ultrason –ou sonification– afin d’obtenir de courts fragments, de l’ordre d’une centaine de nucléotides. On effectue ensuite une PCR non ciblée avec des amorces aléatoires pour amplifier les fragments avant de les séquencer pour obtenir des lectures couvrant l’ensemble du matériel génétique.
La construction d’une table de comptage à partir des lectures est sensiblement plus compliquée que dans l’approche par gène marqueur. Il existe en effet plusieurs manières d’obtenir une table de comptage à partir des lectures: par comparaison de lectures (Maillet, Collet, Vannier, Lavenier, & Peterlongo, 2014; Maillet, Lemaitre, Chikhi, Lavenier, & Peterlongo, 2012), par comparaison des profils en \(k\)-mers (Benoit et al., 2016; Deorowicz, Kokot, Grabowski, & Debudaj-Grabysz, 2015), par classification exhaustive des lectures (Brady & Salzberg, 2009; Kim, Song, Breitwieser, & Salzberg, 2016; Ounit, Wanamaker, Close, & Lonardi, 2015; Wood & Salzberg, 2014), par recensement de gènes marqueurs (Liu, Gibbons, Ghodsi, Treangen, & Pop, 2011; Segata et al., 2012; Truong et al., 2015) ou encore par utilisation d’un catalogue de gènes (Coelho et al., 2019; Kultima et al., 2012; Pons et al., 2010). Nous allons détailler cette dernière méthode, qui nécessite la construction préalable d’un catalogue ou l’utilisation d’un catalogue public (Almeida et al., 2020).
Une fois le catalogue obtenu, chaque lecture est alignée contre celui-ci pour déterminer le gène auquel elle correspond le plus, sur la base de la similarité de séquence. Dans le meilleur des cas, une lecture ne s’aligne que sur un seul gène, mais il arrive dans environ \(10~\%\) des cas qu’elle s’aligne sur plusieurs gènes distincts, par exemple parce qu’elle correspond à un domaine protéique partagé par plusieurs séquences. Dans ce dernier cas, plusieurs procédures sont possibles :
On ne prend pas en compte cette lecture dans le comptage.
Le compte de cette lecture est réparti uniformément entre les gènes (i.e. elle augmente le comptage de chacun des \(n\) gènes sur lesquels elle s’aligne de \(1/n\)).
Le compte de cette lecture est réparti entre les gènes, au prorata de leurs comptages obtenus à partir des lectures non ambiguës (i.e. si elle s’aligne sur les gènes \(G_1, \dots, G_n\) de comptages respectifs \(A_1, \dots, A_n\) dans les lectures non ambiguës, l’abondance du gène \(i\) est augmentée de \(A_i / \sum_{j=1}^n A_j\)).
Contrairement à l’approche par gène marqueur, l’approche non-ciblée sur catalogue de gènes dresse un inventaire fonctionnel du microbiote, où le gène remplace l’OTU comme descripteur de base du microbiote.
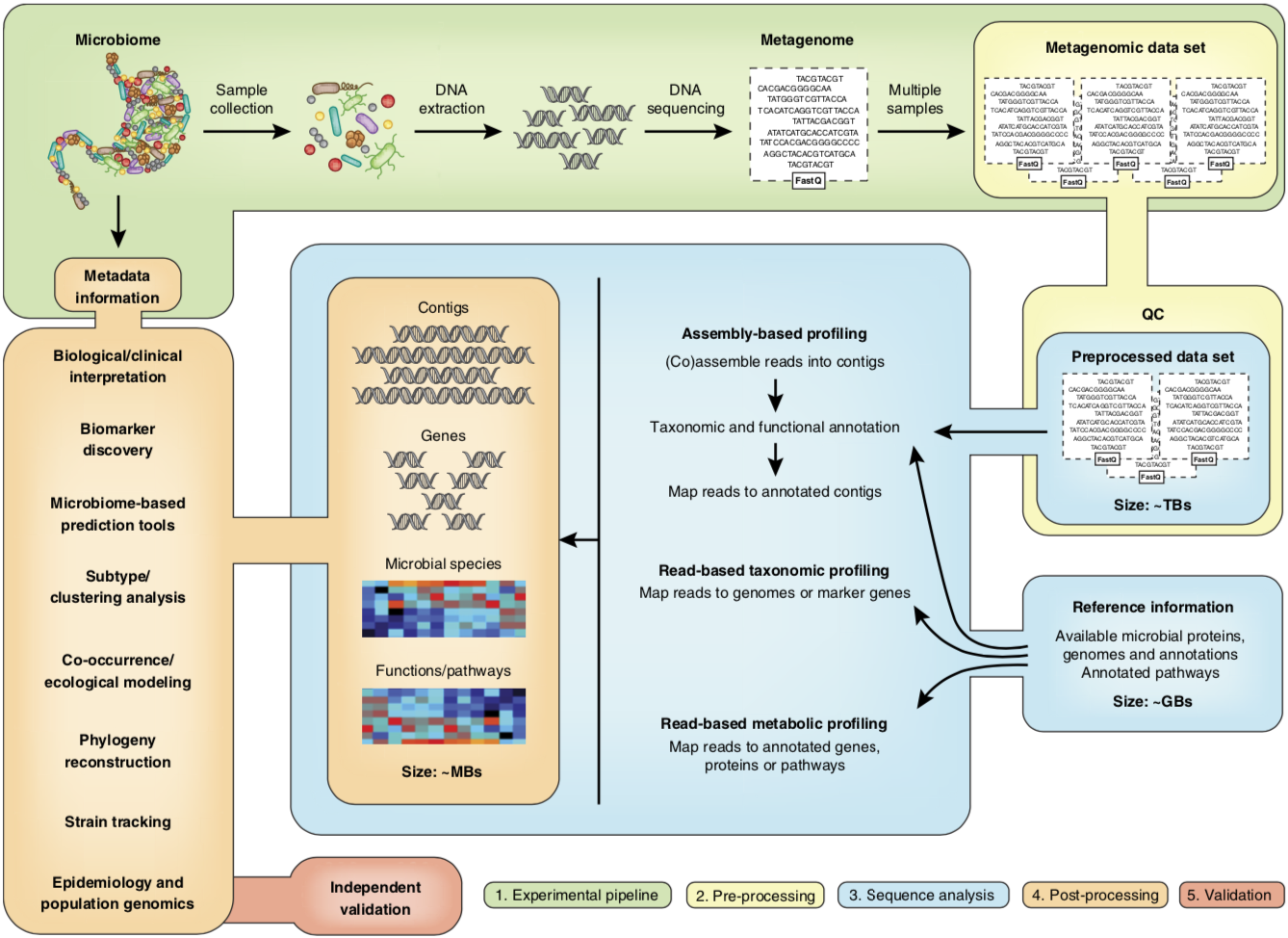
Figure 1.4: Résumé des étapes du séquençage non ciblé, tiré de Quince, Walker, Simpson, Loman, & Segata (2017).
1.2.4 Données métagénomiques
Une fois l’annotation effectuée, on dispose d’une table de comptage, c’est-à-dire du compte des différentes entités considérées dans les échantillons. Dans la suite, on désignera ces entités sous le terme générique taxon, qu’il s’agisse d’espèces, de genres bactériens, d’OTUs, de gènes…
Plusieurs cadres conceptuels existent pour analyser ces données mais les deux plus populaires sont ceux (i) des données de comptage et (ii) des données compositionnelles. Chacun de ces cadres possède des avantages et inconvénients qui lui sont propres et malgré des discussions intenses dans la littérature, (Gloor, Macklaim, Pawlowsky-Glahn, & Egozcue, 2017; Gloor et al., 2016; McMurdie & Holmes, 2014; Vandeputte et al., 2017), il n’existe pas à l’heure actuelle de consensus sur celui à privilégier.
Données de comptage
Par la façon dont elles sont construites, à savoir en comptant le nombre de lectures de chaque taxon dans chaque échantillon, les tables d’abondance sont composées de données de comptage, à valeurs dans \(\NN\).
Une approche naturelle serait de considérer ces comptes comme issus d’une loi de Poisson, dont les masses sont telles que \(\PP{X = k} = \frac{\lambda^k}{k!}e^{-\lambda}\) pour \(k \in \NN\). Cependant, l’espérance et la variance d’une loi de Poisson sont égales, alors que les données observées sont surdispersées : elles possèdent une plus grande variabilité qu’attendue pour une distribution de Poisson (Anders & Huber, 2010; Robinson & Smyth, 2007). Afin de prendre en compte une telle surdispersion dans les données, il est préférable d’utiliser une loi binomiale négative (Zhang et al., 2017), qui peut être vu comme un mélange de loi de Poisson, avec une densité \(\Gamma\) sur le paramètre de la loi de Poisson.
Dans sa définition classique, la loi binomiale négative de paramètres \(n\) et \(p\) compte le nombre d’échecs avant d’obtenir \(n\) succès pour un événement binaire, avec \(p\) comme probabilité de succès. Sa fonction de répartition est alors telle que
\[\begin{equation*} \PP{X = k} = \binom{k+n-1}{n-1} p^n(1-p)^k. \end{equation*}\]
On préférera utiliser ici une autre paramétrisation, celle de moyenne \(m\) et de paramètre de dispersion \(\alpha \geq 0\). Lorsque \(\alpha = 0\), on retombe sur une loi de Poisson. Dans le cas contraire,
\[\begin{equation*} \PP{X = k} = \frac{\Gamma(k+r)}{k!\Gamma(r)}\left(\frac{m}{r+m}\right)^r\left(\frac{r}{r+m}\right)^k \end{equation*}\]
où \(r=\frac{1}{\alpha}\) et \(\Gamma : z \mapsto \int_0^{+\infty}t^{z-1}e^{-t}\dx{t}\) est la fonction gamma.
Une autre particularité des données métagénomiques est leur grande proportion de zéros : les comptages nuls sont surreprésentés et représentent fréquemment \(90~\%\) ou plus des coefficients de la table. Ceux-ci peuvent être des zéros structurels –de nature biologique– où le taxon n’est pas présent dans le jeu de données, ou des zéros d’échantillonnage –de nature probabiliste– où le taxon n’a pas été détecté alors qu’il est présent (mais en faible quantité) dans le jeu de données. Pour modéliser cette fréquence élevée de comptages nuls, on utilise alors des lois avec excès de zéros. Si \(X\) suit une loi sur \(\NN\), on peut créer \(Y\) avec excès de zéros comme suit :
\[\begin{equation*} \left\{ \begin{aligned} \PP{Y = 0} & = p_0 + (1-p_0) \PP{X = 0}\\ \PP{Y = k} & = (1-p_0) \PP{X = k} & \text{pour } k\in \NN^*, \end{aligned} \right. \end{equation*}\]
où \(p_0\) est la proportion de zéros structurels. Si \(X\) suit une loi binomiale négative, \(Y\) suivra alors une loi binomiale négative avec excès de zéros, ou ZINB (pour Zero-Inflated Negative Binomial) (Xinyan, Himel, & Nengjun, 2016).
La somme des comptages des taxons dans un échantillon est appelé profondeur de séquençage et correspond au nombre de lectures produites par le séquenceur pour l’échantillon. La profondeur cible (typiquement \(100~000\) lectures par échantillons) est imposée par le scientifique mais la profondeur effective peut varier d’un facteur \(4\) pour des échantillons avec la même cible (\(50~000\) lectures pour le premier, \(200~000\) pour le deuxième). Il est d’usage d’utiliser un facteur de normalisation pour prendre en compte ces différences et rendre les comptages comparables entre échantillons.
Données compositionnelles
L’autre grand point de vue considère que les comptages sont contraints par la profondeur de séquençage et ne nous permettent donc d’étudier que les abondances relatives (par opposition aux abondances absolues) des différents taxons dans l’échantillon. En pratique, les comptages des \(m\) taxons de chaque échantillon sont divisés par la profondeur de séquençage de l’échantillon pour reconstruire des vecteurs d’abondance relative, à valeurs dans le simplexe \(\mathcal{S}^m = \left\{x \in \RR_+^{*,m}, \sum_{i=1}^m x_i = 1 \right\}\) (Gloor & Reid, 2016).
Ces vecteurs de proportion peuvent ensuite être modélisées comme des tirages de lois de Dirichlet. La loi de Dirichlet \(\mathcal{D}(\alpha)\) sur \(\mathcal{S}^m\) de paramètre \(\alpha = (\alpha_1, \ldots, \alpha_m) \in \RR_+^{*,m}\) a pour densité
\[\begin{equation*} f(x_1, \ldots, x_m) = \frac{1}{\text{B}(\alpha)} \prod_{i=1}^m x_i^{\alpha_i-1}, \end{equation*}\]
où \(\text{B} : \alpha \mapsto \frac{\prod_{i=1}^m\Gamma(\alpha_i)}{\Gamma\left(\sum_{i=1}^m \alpha_i\right)}\) est la fonction beta.
Des approches plus sophistiquées, basées sur des lois hiérarchiques multinomiales Dirichlet, permettent de modéliser en plus le fait que les comptages sont une version bruitée du vecteur de proportion (Holmes, Harris, & Quince, 2012).
\[\begin{equation*} \begin{aligned} p & \sim \mathcal{D}(\alpha), \\ \PP{X_1 = n_1, \ldots, X_m = n_m\mid p} & = \frac{n!}{\prod_{i=1}^m n_i!}\prod_{i=1}^m p_i^{n_i}. \end{aligned} \end{equation*}\]
La transformation en proportions préserve certaines propriétés, par exemple le rang d’un taxon au sein d’un échantillon, mais requiert une attention particulière pour d’autres opérations élémentaires, comme le calcul d’une composition moyenne. Aitchison (1982) est le premier à proposer une géométrie compositionnelle du simplexe, qui diffère de la géométrie euclidienne standard. Dans cette géométrie, le simplexe possède ses propres opérations internes de perturbation \(\oplus\) ou de composition \(\odot\) ainsi que sa propre distance \(d_a\) définie par
\[\begin{equation*} d_a(x, y) = \sqrt{\frac{1}{2m}\sum_{i=1}^m\sum_{i=1}^m\left(\ln\left(\frac{x_i}{x_j}\right)-\ln\left(\frac{y_i}{y_j}\right)\right)^2} \text{ pour } x,y\in\mathcal{S}^m. \end{equation*}\]
Un point régulièrement évoqué, que la géométrie d’Aitchison corrige, est la présence de corrélations négatives fallacieuses dans le jeu de données normalisé par la somme. Pour s’en convaincre, on peut considérer \(2\) variables indépendantes \(X\), \(Y\) et constater que \(p_1 = \frac{X}{X+Y}\) et \(p_2 = \frac{Y}{X+Y} = 1 - p_1\) sont corrélées négativement. Aitchison propose plusieurs transformations, collectivement appelées xlr, pour pour plonger le simplexe dans l’espace euclidien standard et pouvoir ainsi appliquer les méthodes d’analyse multivariée standards aux données compositionnelles (Pawlowsky-Glahn, Egozcue, & Tolosana Delgado, 2007).
- Le ratio logarithmique additif (additive log-ratio) (Aitchison, 1986)
\[\begin{equation*} \text{alr}: x \in \mathcal{S}^m \mapsto \left(\ln\left(\frac{x_1}{x_m}\right), \ldots, \ln\left(\frac{x_{m-1}}{x_m}\right) \right) \in \RR^{m-1}. \end{equation*}\]
Cette transformation souffre de deux problèmes : elle ne conserve pas les distances entre le simplexe et \(\RR^{m-1}\) et nécessite d’utiliser un taxon de référence (Albarède, 1996; Pawlowsky-Glahn et al., 2007).
- Le ratio logarithmique centré (centered log-ratio) (Aitchison, 1986)
\[\begin{equation*} \text{clr}: x \in \mathcal{S}^m \mapsto \left(\ln\left(\frac{x_1}{g_m(x)}\right), \ldots, \ln\left(\frac{x_{m}}{g_m(x)}\right) \right) \in \RR^{m} \end{equation*}\]
où \(g:x\mapsto \sqrt[m]{\prod_{i=1}^m x_i}\) est la fonction de moyenne géométrique. Bien que cette transformation conserve les distances et ne nécessite plus d’utiliser un taxon de référence, le simplexe est plongé dans un sous-espace vectoriel de dimension \(m-1\) de \(\RR^m\) défini par \(\{y \in \RR^m: y^T 1_m = 0\}\). Autrement dit, la somme des coordonnées du projeté doit être nulle.
- Le ratio logarithmique isométrique (isometric log-ratio) (Egozcue, Pawlowsky-Glahn, Mateu-Figueras, & Barcelo-Vidal, 2003).
\[\begin{equation*} \text{ilr}: x \in \mathcal{S}^m \mapsto \Psi^T \text{clr}(x) = \left(y_1, \ldots, y_{m-1}\right) \in \RR^{m-1} \end{equation*}\]
où \(\Psi\) est une base orthonormée quelconque du sous-espace vectoriel \(\text{clr}(S^m) = \{y \in \RR^m: y^T 1_m = 0\}\). Cette transformation conserve les distances et nécessite une base adaptée. Un choix classique de base est donné par la matrice de Helmert privée de sa première ligne (et représentée ici pour \(m = 4\))
\[\begin{equation*} \Psi^T = \begin{pmatrix} 1/\sqrt{2} & -1/\sqrt{2} & 0 & 0 \\ 1/\sqrt{6} & 1\sqrt{6} & -2\sqrt{6} & 0 \\ 1/\sqrt{12} & 1\sqrt{12} & 1\sqrt{12} & -3\sqrt{12} \end{pmatrix}, \end{equation*}\]
pour laquelle on obtient
\[\begin{equation*} y_{i-1} = \frac{1}{\sqrt{i(i+1)}} \ln\left(\frac{x_i}{\left(\prod_{j=1}^{i-1} x_j\right)^{\frac{1}{i-1}}}\right) \text{ pour } i \in [\![2,m]\!]. \end{equation*}\]
Cette base produit des coordonnées \(y_i\) facilement interprétables : \(y_i\) mesure la balance entre \(x_i\) et la moyenne géométrique de \(x_1\) à \(x_{i-1}\).
Si l’on dispose de l’arbre phylogénétique des taxons, d’autres contrastes interprétables peuvent également être utilisés, par exemple la balance entre le sous-arbre gauche et le sous-arbre droit d’un nœud de l’arbre (Silverman, Washburne, Mukherjee, & David, 2017).
Enfin, Xia, Sun, & Chen (2018) proposent une autre base, qui donne les coordonnées suivantes.
\[\begin{equation*} y_i = \frac{1}{\sqrt{i(i+1)}}\ln \left(\frac{\prod_{j=1}^i x_j}{(x_i + 1)^i}\right) \text{ pour } i \in [\![1,m-1]\!]. \end{equation*}\]
Les transformations xlr ne tolèrent pas des proportions nulles. La solution généralement adoptée consiste à ajouter un pseudo-compte de \(1\) (ou \(1/2\)) à tous les taxons avant de calculer les proportions. Cette solution, si elle permet en pratique de s’abstraire des zéros lors des calculs, présente néanmoins l’inconvénient majeur d’induire des pics dans la densité des coordonnées transformées sans pour autant permettre de gérer explicitement les zéros structurels.
1.3 Jeux de données
Nous présentons ici les jeux de données utilisés dans ce manuscrit. Chaque jeu de données porte le nom du premier auteur de l’étude dont il est extrait. Tous sont disponibles dans le matériel supplémentaire de l’article d’origine ou dans le package R {curatedMetagenomicData} qui met à disposition de manière homogène des jeux de données de métagénomique (Pasolli et al., 2017; R Core Team, 2020). Afin de limiter le bruit lié aux taxons très peu présents dans les jeux de données, ceux-ci pourront être retirés si leur prévalence (i.e. le pourcentage d’échantillons dans lesquels le taxon est présent) est en dessous d’un certain seuil.
Brito
Brito et al. (2016) comparent une cohorte de \(81\) Nord-Américains urbains avec une cohorte de \(171\) Fidjiens ruraux en utilisant à la fois du séquençage 16S (que nous utiliserons) et du séquençage non ciblé. Leurs travaux ont montré que la variation du régime alimentaire se reflète dans la variation du potentiel fonctionnel des gènes du microbiote, les Fidjiens ayant par exemple plus de gènes spécialisés dans la dégradation de l’amidon. En ne gardant que les \(112\) adultes fidjiens de cette cohorte pour former un groupe d’échantillons homogènes, il reste \(77\) OTUs.
Chaillou
L’étude de Chaillou et al. (2015) s’intéresse au microbiote alimentaire de produit carnés (bœuf haché, veau haché, merguez de volaille et dés de lardons) et de produits issus de la mer (filet de cabillaud, crevette, filet saumon et saumon fumé) pour étudier le rôle du microbiote dans l’altération de l’aliment. Les \(64\) échantillons, répartis uniformément entre les différents aliments, ont mis en évidence une perte de diversité microbienne concomitante à l’altération et ont permis d’identifier des espèces associées à une altération précoce. En ne conservant que les taxons ayant une prévalence supérieure à \(5~\%\) de prévalence, on conserve \(499\) OTUs dont \(97\) assignées à l’embranchement des Bacteroidetes.
Chlamydiae
Seule exception à notre terminologie, le jeu de donnée Chlamydiae constitue une sous-partie du jeu de données construit par Caporaso et al. (2011). Ce dernier contient \(26\) échantillons répartis au sein de \(8\) environnements très différents (selles, bouche, eau, sol, sédiments, océan, eau douce calme et eau douce vive) pour étudier la diversité microbienne globale et calibrer l’effort de séquençage nécessaire à une bonne caractérisation. Le sous-jeu de données est restreint aux \(21\) OTUs assignées à l’ordre des Chlamydiales et a servi d’exemple pour {StructSSI} dans Sankaran & Holmes (2014).
Ravel
Le jeu de données présenté dans Ravel et al. (2011) concerne le microbiote vaginal de \(396\) femmes nord-américaines, n’ayant pas atteint la ménopause, issues de différents groupes ethniques et sujettes ou non à des vaginoses. Le séquençage par gène marqueur 16S a permis d’identifier cinq archétypes de communautés microbiennes: quatre d’entre eux sont dominés par une espèce de Lactobacillus qui acidifie le milieu et empêche le développement de bactéries responsables de vaginose, le dernier correspond à une diversité bactérienne élevée et est associé à des risques accrus de vaginose. \(40\) genres différents, de prévalence supérieure à \(5~\%\) sont présents dans ce jeu de données.
Wu
Wu et al. (2011) se sont intéressés aux relations entre microbiote et régime alimentaire, parmi lesquelles l’importance de la consommation d’alcool. Bien qu’un changement de régime alimentaire pendant une courte durée (\(10\) jours) ait un impact significatif sur le microbiote, son ampleur reste modeste. Ce jeu de données, qui comprend \(98\) échantillons répartis à égalité entre sujets à faible et forte consommation d’alcool, contient \(400\) OTUs.
Zeller
Le jeu de données issu de Zeller et al. (2014) contient \(42\) patients ayant un adénome, \(91\) patients ayant un cancer colorectal et \(66\) volontaires sains. Le but de l’étude est d’étudier les associations entre la composition du microbiote et le statut du patient, notamment pour trouver des biomarqueurs de la maladie. Tous les échantillons ont été caractérisés par l’approche gène marqueur (à l’aide du gène 16S) et par l’approche non-ciblée. En ne conservant que les taxons ayant une prévalence supérieure à \(~5\%\), l’approche gène marqueur a permis d’identifier \(119\) genres différents tandis que l’approche non-ciblée a identifié \(878\) MSPs –une autre entité métagénomique (Plaza Oñate et al., 2018).
| Jeu de données | Microbiote | Rang | Taxons | Échantillons |
|---|---|---|---|---|
| Brito et al. (2016) | Intestinal | OTU | 77 | 112 |
| Chaillou et al. (2015) | Alimentaire | OTU | 499/97 | 64 |
| Chlamydiae
(Caporaso et al., 2011) |
Varié | OTU | 21 | 26 |
| Ravel et al. (2011) | Vaginal | Genre | 40 | 396 |
| Wu et al. (2011) | Intestinal | OTU | 400 | 98 |
| Zeller et al. (2014) | Intestinal | Genre | 119 | 199 |
| Zeller et al. (2014) | Intestinal | MSP | 878 | 199 |